
Sarepta (SRPT) had a great run last week, a kick off following MS fireside chat, closing at $152.76 the highest “close” since June 21st (highest 52W close 06/20 was $153.69) on nice volume, but the highest “Weekly-Close” in the weekly chart (Close & not highest price point). Looking for a continuation next week, while the target is breaking out the 52Weeks-High $176.50 as well, anticipating Doug’s next presentation at Janney Montgomery Scott Healthcare Conference on Tuesday 09/18/2018 at 8:15 AM EDT, where we expect a rehash of the MS fireside chat, & maybe news about the clinical-Hold lifting by the FDA.
But the next important milestone is the WMS-2018 conference where Dr. Mendell will take part at the evening symposium as confirmed by SRPT’s CEO and will present the biopsy results from the fourth patient, and more important functional data from the 4 kids, where the first patient will have full 10 months data since his gene-therapy was back on January 04th, 2018.
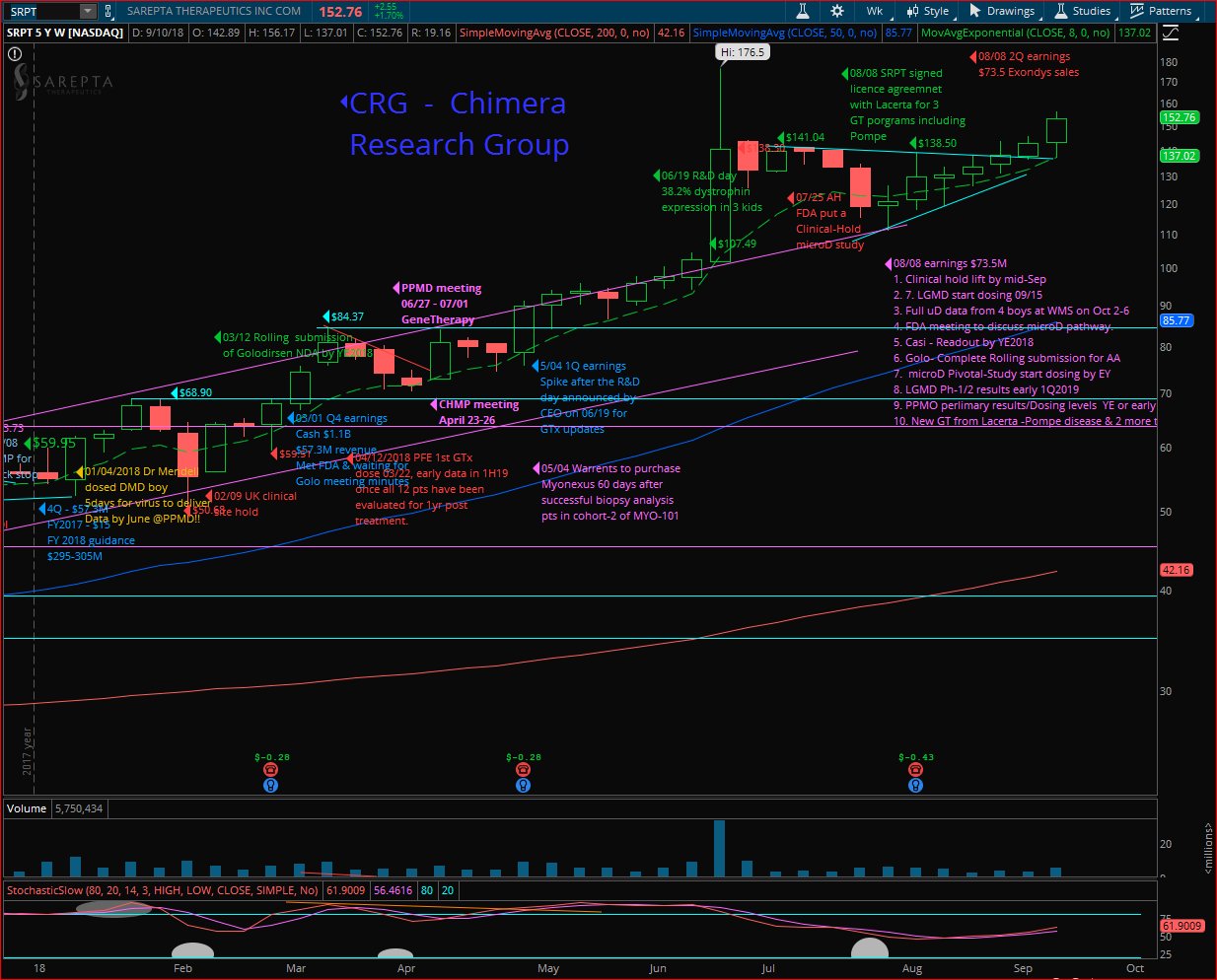
Golo rolling submission is going well and will submit the final NDA by YE-2018, and the FDA review by Mid-2019, while Casi with data by YE-2018 and if the results are good, we will target submitting by Mid-2019 and approval by YE-2019 to 1Q-2020. Then we will cover over 30% of the DMD community with Exondys-51, Golo & Casi.
Gene therapy had a very exciting data, and SRPT is building a GT power house, will have their own lab running up, within 2.5 years will have a pipeline of around 30 GT programs!!!
Questions & Answers:
Q: SRPT had 3 patients data in microD with biopsy data on 06/19, walk us through the process.
A: Doug: On 06/19 we had 4 patients dosed, but the 4th patient had no biopsy yet, we had biopsy from the first 3 patients to present on the R&D day, and Dr. Mendell will update on the WMS-2018 on 10/03 4th biopsy & as well as I am quite confident he will provide some insight on what he’s seeing from a functional perspective across all 4 boys. The data on 06/19 was extraordinary expression level from 3 months biopsy data. highest ever WB level, we had also about 1.6 genome copies per muscle cells & 120% expression in the heart.-Cardiac muscles. We saw 70% positive fiber in children, also significant drop in CK levels that associated with muscle damage (near 90%), we have to dose more kids, to watch them, get to the FDA & get confirmation of the registration study pathway.
A: I think it’ll be a little better than anecdotal first, as 1st patient dosed on 01/04/2018 so it’s close to a year of data. And I’m quite confident he will present data versus baseline. It’s also a little better than anecdotal in the sense that Dr. Mendell is an extraordinary clinician who is known this disease unfortunately for decades and decades, and so he can provide his insight into what he’s seeing and what that might mean both quantitatively, but also qualitatively. In the end, we have to take all of this information, show the FDA to discuss the clinical pathway, and then go into registration trial and sort of firm this all in a well-controlled clinical trial.Q: Clinical hold, are you on track with your plan & how you will produce the material without fragment?
A: We were placed on clinical hold recently as a result of the non-producing fragment that was found in a plasmid from a third party supplier to our GMP manufacturer, Nationwide. That was because there was research grade plasmid that was used, and so we all know research grade plasmid is a very well used as it is a standard. I think probably until our clinical hold, it was the standard for early gene therapy.
I think the FDA is increasing the standard, and probably in part because they are actually increasing their view on how fast one can move and the way we ought to design our clinical trials. Now to do that, we really need to get in front of the FDA and have a robust discussion with the FDA. Show them our plans for this trial, and get their confirmation that these plans are appropriate and that they could form the basis of an approval. And our goal is to do that hopefully by the end of November, perhaps into early December.
We already responded to the clinical hold. We certainly hope that it was a complete response. That was certainly our goal. Fairly straightforward response. We committed to using GMP sourced material in the future, and audited the third party plasmid supplier to confirm that the third party plasmid supplier has GMP sourced material and abides by the principles of GMP. And we did that, and they do. So it is certainly our hope that unless there is some additional question about which we are not aware, that we would be off clinical hold in plenty of time for us to meet our goals, which is to begin dosing these children in what we hope to be a registration trial by the end of this year.
Q: any more patients will be dosed in the current Cohort B?
A: We are going to focus on what we hope is a pivotal arm, so we’re not going to dose additional children (in Cohort-B). I think the point of the original dosing was a proof of concept, which was the way people were thinking of things previous to the FDA’s recent guidance. And I think given the data we have seen, we have certainly I think satisfied the POC. So we really want to move into what we hope to be a pivotal trial and move as fast as possible to bring this therapy to the community.
Q: You outlined how the pivotal study will look like?
A: Cohort-C with 24 patients placebo controlled 1:1 study, we hope it will be a registration trial, and all this done before the FDA issue guidance on rare disease & it’s aligned with our goal.
Q: I guess the next step, which is an area of debate obviously, is manufacturing scale up, your ability to scale appropriately and be ready for clinical supplies so you can meet your sort of aggressive time schedule related to filing. So maybe just walk people through where you are now, what you are doing with Brammer, and what the steps are in between that you have to meet to get there.
A: We have supply from NCH for the clinical study, Braemar manufacturing is for the commercial supply & will be ready for launch, we also learn from AVXS approach. and will have tech transfer from NCH to Braemar. working on scale up & our goal from timeline perspective to start dosing kids by end of this year (subject of discussion with FDA) for this study, 24 patients (12 drug & 12 Placebo) & follow them for a year, and those placebo kids will crossover to actual therapy, when unblinded & give these kids therapy, we will be in a position of a commercial supply to dose those kids & this will give us to use data to bridge & submit data the BLA & get approval within 24 months.
A: The fact there are 2 other companies make us work harder & faster, unlike others we have Rh74 capside that cross the BBB which is a significant value to us & we have a lower level of screened antibody around 15% which is much lower than the other programs. we also use a different promoter that designed to over expressed in the heart with over 100% it’s very important.
Q: Golo & Casi timeline?
A: it was a big moment for us in Feb 2018 when we sat with the FDA & talked about the ability to use dystrophin as a surrogate marker for approval of Golo. Ex-53 will be ending the rolling submission by year end & submit the NDA & target approval mid 2019…then Casi is another 8% of DMD community, we met with the FDA as well to do an interim look blinded on dystrophin production & the FDA was supportive & we will be looking at those biopsies by year end, if we see a good dystrophin production as preclinical models, then we will submit by mid 2019 & target approval by YE2019 or early 1Q2020
A: We have 6 PPMO programs, the 1st is Exon-51 single dose study, we will get dosing insight by 1Q next year. and IND enabling on 5 programs as well.
Q: LGMD what is the study design
A: LGMD is the same capside & same prompter as microD, we are very excited about it, we’ll dose this year in 3 patients, first patient will be dosed this month (Sep 15 IMO) and data of 3 patients in 1Q next year to give us insight, 2E program is a self complementary vector so we will use a very low doses to get a high expression levels & because of the efficiency we will look at biopsy in 2 months instead of 3 months.. so will move fast.
LGMD is about the same size of the DMD community in patients population so this is a very significant opportunity.












